How To Select The Right Molecules To Make And Test
Introduction
In small molecule drug discovery, the question of which molecules to make and test in the next experimental cycle is undoubtedly one of the most consequential. Advanced program molecules typically require custom synthesis and each chemist can only make a few molecules per week. Picking the wrong set of molecules to synthesize can set a program back for many months or even years, increase costs significantly, and ultimately lead to the high failure rate that is endemic to drug discovery. Indeed, the ultimate question where AI intersects with experimental reality is what is the optimal small set of compounds to synthesize and test?
Following hit discovery, the design workflow involves generating compounds starting from a desirable hit or lead and using a variety of predictive models to estimate potency, ADME, and other properties. The final step is to then select which compounds (and how many) to synthesize from the large list of profiled molecules. Common selection approaches include rank-ordering the compounds based on a combined score (this is called Greedy) or clustering and selecting the top few compounds (based on the combined score) from each cluster to ensure chemical diversity (this is called Cluster Greedy).
It is well known that neither of these selection strategies (also referred to as acquisition functions) are ideal for selecting a batch of compounds and assaying them in parallel. At best, they hedge the resulting selections by factoring in compound diversity, but do not account for a model’s joint predictive uncertainty.
A standard and effective approach to uncertainty quantification in neural networks is to train an ensemble of models (e.g., K models) on different splits of a given property dataset, and estimate uncertainty based on the resulting K predictions per molecule. This approach works well for lightweight models that are quick to train, but is too costly for today’s massive models, such as potency models based on Pairformers.
We solve this problem by pairing a cost-effective method to quantify model uncertainty (Epinets) with a preferred acquisition function for single properties (expected maximum or EMAX). When combined to power EMMI Select, this approach provides a 3x reduction in time and cost to improve molecule potency to a desired level in retrospective benchmarking. Early prospective testing in Terray’s programs shows similar levels of efficiency that are enabling us to solve difficult challenges and we are working to extend this method to all of the relevant beyond-potency properties essential to small molecule drug discovery. We believe this approach will have a profound impact across the industry.
Epinets Cost-Effectively Quantify Model Uncertainty
At Terray Therapeutics, Experimentation Meets Machine Intelligence through our EMMI platform (see companion insight post), as our full-stack AI platform leverages proprietary experimental hardware and a highly-automated lab to drive a rich pipeline of internal and partnered programs. A key component of our predictive modeling capabilities is Terray’s global potency prediction model (TerraBind), which is trained on both public datasets and our proprietary data which covers the dark, unexplored areas of chemical space. In every cycle, pairing EMMI’s Generative AI with TerraBind produces thousands of molecules for potential synthesis and testing.
 Figure 1: An exemplar potency distribution provided by TerraBind-Epinet
Figure 1: An exemplar potency distribution provided by TerraBind-EpinetTerraBind is trained on such a scale of data that re-training it 100s of times to quantify uncertainty in its predictions is impractical. A solution to this challenge is Epistemic Neural Networks (Epinets). These provide high-quality joint predictions and uncertainty estimates, allowing us to seamlessly incorporate them into a selection workflow. Moreover, unlike in deep ensembles which require storing copies of parameters, this approach requires training TerraBind only once and admits efficient and easily parallelizable batched inference. For example, Epinets can be used to efficiently provide a potency distribution per molecule, as shown in Figure 1.
Pairing Epinets with a Parallel Acquisition Function Leads to a 3x Reduction in Time and Cost
With an efficient approach for joint uncertainty quantification in hand, the next step was to pair the TerraBind-Epinet with an acquisition function that is reflective of the goals in hit-to-lead and lead optimization. For the specific design goal of maximizing potency in our programs, we use the expected maximum (EMAX), a simple, parallel acquisition function that hedges between compounds selected within a batch by penalizing inferred correlations between them. These basic acquisition functions are easily adjusted to address other concerns which realistically emerge while prosecuting programs: maintaining potency while avoiding anti-targets, improving ADME, etc. Figure 2 shows an example workflow of leveraging Epinets + EMAX to determine the optimal size and composition for a batch of molecules to select for synthesis and testing.
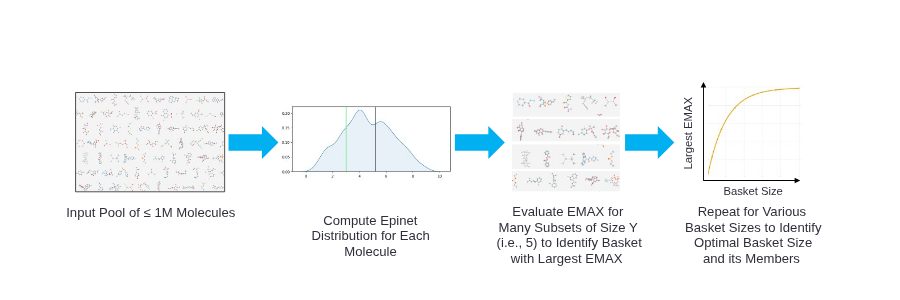 Figure 2: A design workflow that leverages Epinets + EMAX to determine the optimal number of molecules to select for synthesis.
Figure 2: A design workflow that leverages Epinets + EMAX to determine the optimal number of molecules to select for synthesis.Retrospective Benchmarking: EGFR Inhibitors
To benchmark the impact of Epinets + EMAX selections on improving the efficiency of potency optimization, we conducted a retrospective study using a dataset of literature EGFR inhibitors which was augmented with inactive decoys to better simulate the distribution of potency in a real-world setting.
The goal for the study was to identify the most potent compound in a set of ~50,000 molecules. The following process was repeated 25 times for each method and the results averaged to ensure convergence:
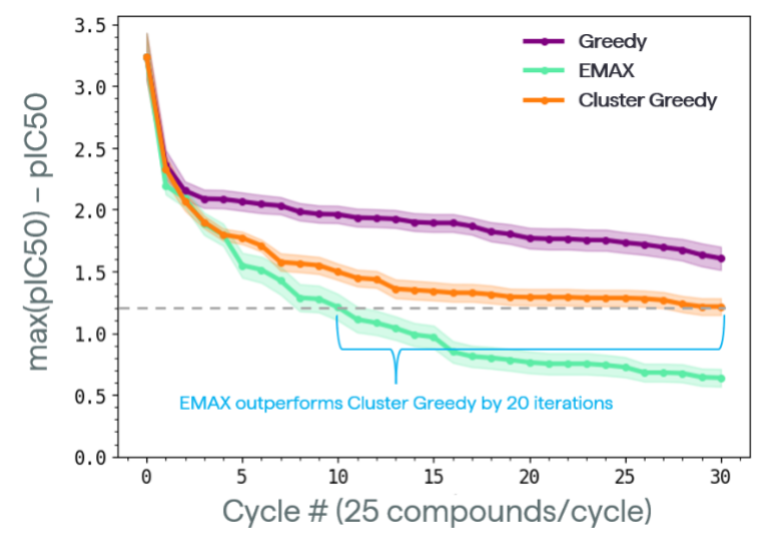 Figure 3: Retrospective analysis of a dataset of EGFR inhibitors.
Figure 3: Retrospective analysis of a dataset of EGFR inhibitors.- For the first cycle, randomly choose 100 compounds
- Re-train the predictive model with the IC50s of these compounds
- Select the next 25 compounds based on the associated selection method (Greedy, Cluster Greedy, or EMAX)
- Repeat this for 30 iterations, simulating a series of DMTA cycles wherein around 10-20 chemists are making molecules
The results are shown in Figure 3. We were encouraged by how much more efficient the EMAX approach was relative to commonly used strategies like Greedy and Cluster Greedy. This can be seen both by the fact that Epinets + EMAX reached the same potency as the average final performance of the Cluster Greedy approach 3x faster, as well as the fact that at the end of the 30 cycles, Epinets + EMAX provided a compound 4x better than Cluster Greedy and 10x better than Greedy in terms of IC50.
Early Prospective Results: Virtual Screening And Program Progression
As soon as the Epinets + EMAX methodology was finalized, we ran a prospective validation by using it to make the final compound selections from a virtual screen of more than 10 billion compounds to find hits for 14 very difficult targets. Despite the fact that most of these targets have no disclosed small molecules in the public literature, Epinets + EMAX selected an average of only 17 compounds per target to identify an active compound for more than 90% of the targets. Furthermore, for half of the targets, the best compound was at least 10 μM, and for two of the targets, the best compound was under 1 μM – often a meaningful program inflection point. The results are summarized in Figure 4.
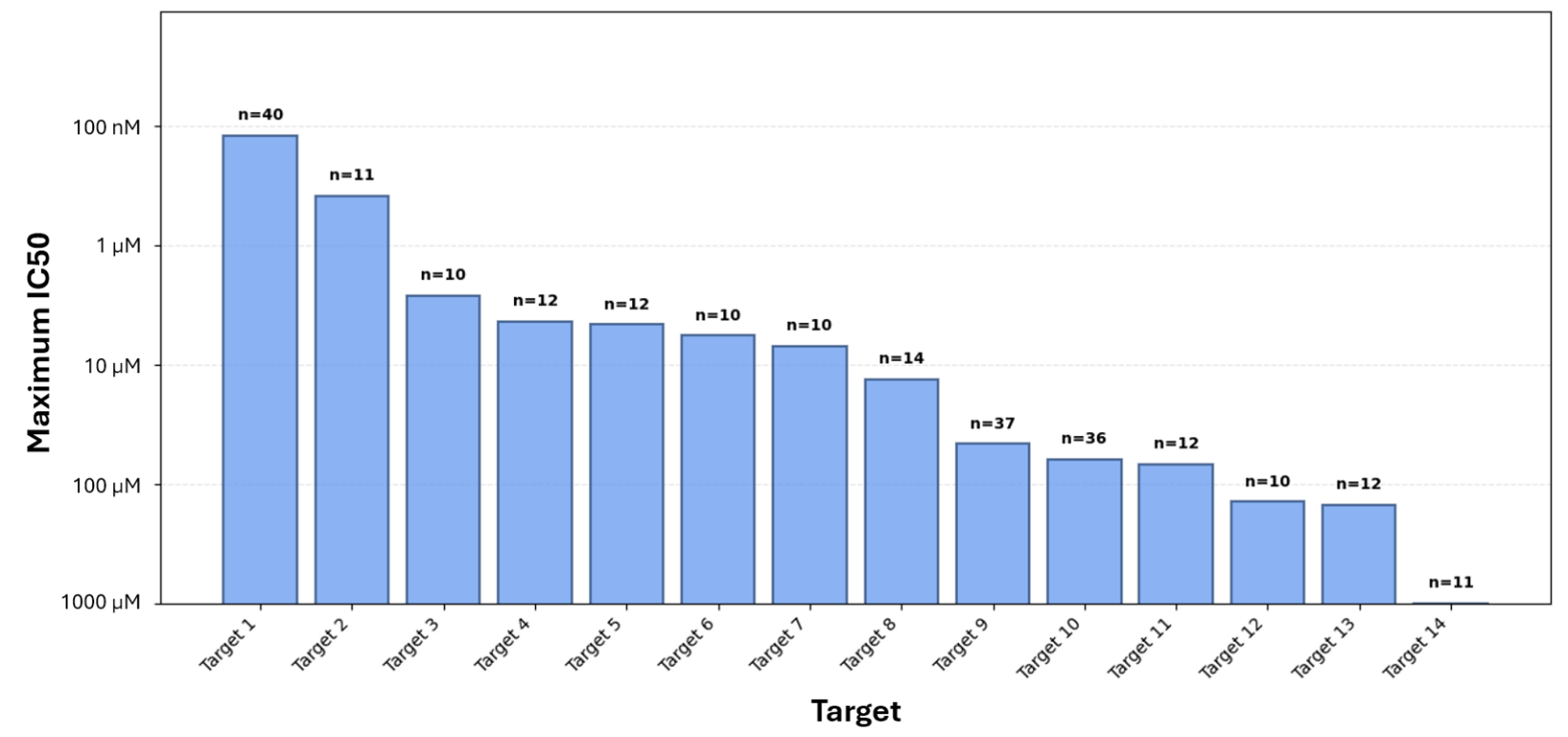 Figure 4: Results of a virtual screen that used Epinets + EMAX to make the final compound
Figure 4: Results of a virtual screen that used Epinets + EMAX to make the final compound To further demonstrate the prospective utility of this method, we chose one of our most challenging targets that had just entered hit-to-lead and needed a major boost in potency. Epinets + EMAX was used to select a batch of 76 compounds for a parallel-synthesis approach, and 80% of the selected molecules provided the desired improvement in potency. If you are interested in learning more details about how to enhance selections with Epinets + EMAX, read our latest manuscript or check out our GitHub, where we introduce a framework to improve the quality of an Epinet’s joint predictions by pretraining on synthetic data.
What’s Next?
Making optimal decisions per DMTA cycle is a very challenging goal, and this work puts us one step closer to consistently choosing the right molecules for the right reasons at the right time. While we are encouraged by the performance of Epinets + EMAX in both retrospective benchmarking and early prospective use, optimal selections for potency solves for just one of many relevant property objectives. The next step is to incorporate additional important properties such as ADME, synthesis/assay cost, and synthesis/assay time, and then extend these selections beyond single-cycle optimizations to multi-cycle. With these additional capabilities, we can make even better decisions that reduce preclinical cycle time and cost, enabling more efficient discovery and increasing the likelihood of clinical success.