Introducing EMMI Predict: TerraBind
One of the holy grails of computational drug discovery has been the ability to accurately predict the binding affinity between a small molecule and a protein target. Rapid progress has been made in the post-AlphaFold era, with models that infer potency from generative all-atom structure models. Simpler models based on ligand-only representations and docking have been effectively rendered obsolete, and structural transformer-based approaches now compete with free-energy models at a tiny fraction of the cost.
At Terray, we’ve developed and integrated our own predictive models deeply into our internal and partnered programs and learned that the architecture used throughout the field is limiting for drug design. AlphaFold and its open-source co-folding frameworks (Boltz-1, NeuralPlexer, SeedFold, OpenFold) focus on the protein structure prediction problem. While this was the original grand challenge, the connection between structure prediction and actual potency isn't direct. Modern “universal potency models” represent the current frontier, utilizing all-atom diffusion to derive both pose and potency. However, while this paradigm yields high-fidelity, all-atom structures, the computational overhead of diffusion is substantial. This limited inference throughput constrains the searchable chemical space, diminishing the model’s practical utility for prioritizing candidates during synthesis selection. Especially today, there is a growing need to evaluate thousands to millions of small molecules at all stages of the discovery process: hit identification, hit-to-lead, and even lead optimization.
This raises a critical question: Do we really need a human-interpretable, all-atom pose (e.g., via diffusion) to achieve accurate potency predictions?
Our hypothesis was “no.” Poses may help with ideation, but if the goal is to screen millions of compounds and rank them for potency, a visualization-quality pose for every single input molecule is unnecessary. You need speed, you need ranking accuracy, and you need error quantification – an often-overlooked but practical necessity for moving programs forward quickly and efficiently.
Today, we are introducing EMMI Predict: TerraBind, a new model that retains the rich structural information provided by protein-ligand interactions without the computational bottleneck of diffusion, and enables the uncertainty quantification necessary for driving decisions in practical drug discovery processes. To read a detailed account of the architecture and performance of TerraBind, see our arXiv paper.
The TerraBind Approach: Potency Over Pose
At Terray, we conceptualize potency prediction as fit-for-purpose tools. For large scale virtual screens, you have sequence-only models. Our in-house model is TerraBind-Seq, which combines protein representations from ESM-2 with molecular representations from COATI-3, our latest chemical foundation model, which has been pretrained on over one billion small molecules. These kinds of models are fast but lack explicit structural context. For ideation on a few high priority molecules, you have diffusion models (like Boltz-2), which are structurally rich and produce all-atom poses, but too slow for the typical large-scale chemical space evaluations that accompany small molecule design campaigns.
For everything in between, TerraBind-Struct (TerraBind) hits the sweet spot. It learns rich structural representations at a coarse-grained level (protein Cβ atoms and ligand heavy atoms only) within a Pairformer Trunk, then maps those representations directly to binding affinity. Because we bypass diffusion entirely — no iterative denoising, no all-atom coordinate generation — we unlock a 26x speedup without sacrificing the structural information that drives accurate potency prediction. The results are a massive leap forward in efficiency without sacrificing rigor. And, when combined with our TerraBind-Seq model (which is significantly faster than TerraBind-Struct), this funneled approach allows us to routinely and accurately evaluate chemical spaces that are in the billions of molecules.
1. Speed: Unlocking Million-Scale Screening
By eliminating the diffusion bottleneck, TerraBind-Struct achieves a dramatic reduction in inference time.
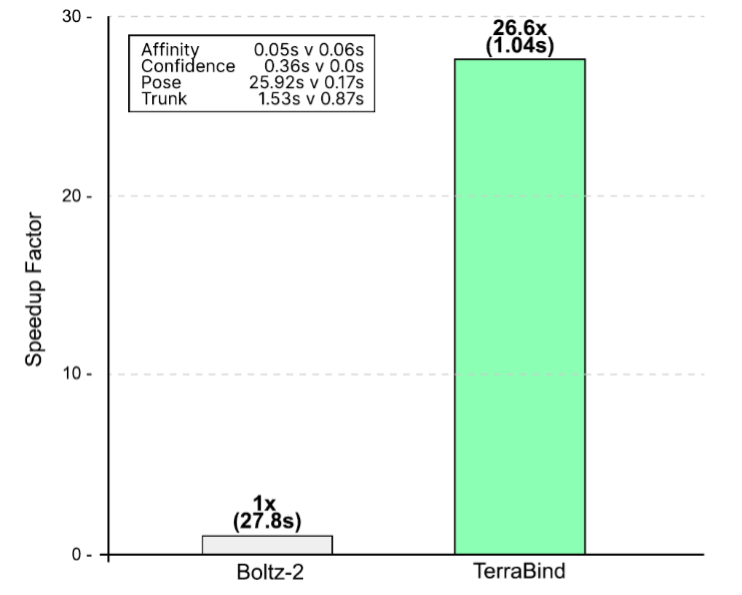
We observe a 26x speedup compared to Boltz-2 (end-to-end inference time per complex on a single A6000 GPU using 196 tokens, 10 samples, and NVIDIA cuEquivariance kernels). This efficiency gain transforms the model’s utility.
Instead of reserving high-accuracy structure-based scoring for a few thousand filtered hits, you can now screen chemical spaces 26 times larger using the same resources – or screen the same space at 96% lower compute cost.
2. Potency: Higher Accuracy for Ranking
Counterintuitively, bypassing the diffusion step doesn’t hurt accuracy. By removing the diffusion head, we force the Pairformer to fully resolve structural features rather than
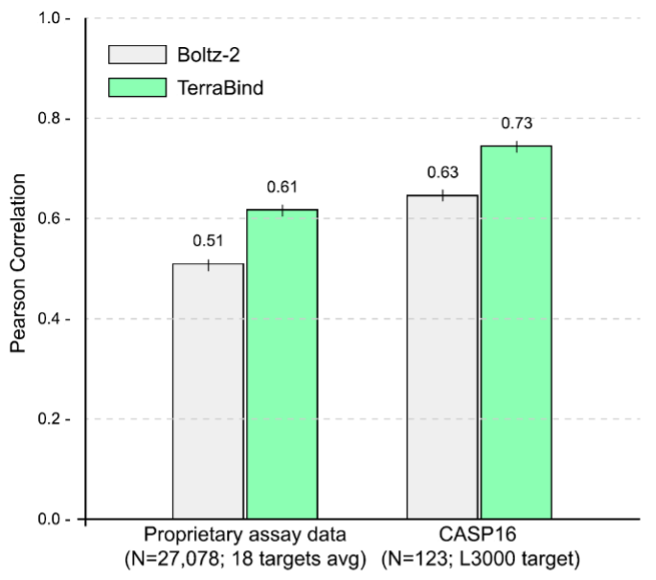
offloading refinement to a downstream generative step. Combined with a strong ligand encoder (COATI-3) and expanded synthetic protein-ligand structural training data, the resulting model outperformed the diffusion baseline.
On two independent held-out test sets, TerraBind demonstrated a 16–20% improvement in Pearson correlation relative to Boltz-2. These include the public CASP16 binding affinity challenge and a proprietary dataset of over 25,000 assay readouts spanning 18 distinct targets. In the latter, we found that TerraBind outperformed Boltz-2 in 15 out of the 18 targets.
Notably, a few days after our preprint appeared on arXiv, Isomorphic Labs announced their latest potency model, IsoDDE. On the same CASP16 benchmark where we surpass Boltz-2 accuracy by 16%, TerraBind matches the performance of IsoDDE. We cannot directly compare IsoDDE and TerraBind on speed/cost and affinity uncertainty quantification, as neither is described for IsoDDE, but we assume that IsoDDE retains the diffusion step that limits scaling due to speed/cost, and lacks uncertainty quantification for affinity.
3. Structure: Retaining Physical Relevance
For any significant architectural changes, such as the ones that accompany TerraBind, it is very important to ensure that the gains in performance are still grounded in physical reality. We assess this via pose accuracy, which gives us a way to evaluate the quality of TerraBind’s structural features (Pairformer) and to produce interpretable binding modes. To generate 3D coordinates from the output predicted by the Pairformer, we developed a simple optimization routine that requires no learnable parameters.

While TerraBind sacrifices the granular, all-atom detail found in diffusion outputs, it consistently captures the coarse structural features essential for reliable pose prediction and affinity. We show performance on a popular metric (% of ligands with RMSD < 2Å) across 2,687 complexes in the Runs N’ Poses dataset, comparing four models with the same training cutoff (September 30, 2021):
- Boltz-1: Full diffusion-based pipeline
- Boltz-1 Trunk: Boltz Pairformer features evaluated directly with our optimization routine, without diffusion
- TerraBind Pocket: A streamlined variant that focuses only on the binding site, using either a TerraBind-predicted pocket or a known pocket from drug discovery programs
- TerraBind: Our coarse-grained structural model
TerraBind matches the full diffusion pipeline while significantly outperforming Boltz-1 Trunk – evidence that our Pairformer learns richer structural representations, and that coarse-grained geometry is sufficient for accurate pose prediction.
Beyond Greedy: Uncertainty Quantification for Optimal Selections
In hit-to-lead and lead optimization campaigns, a purely greedy selection approach (picking the compounds that rank highest for a given metric) often underperforms in practice
since it insufficiently samples chemical space and does not hedge against predictive uncertainty.
TerraBind natively leverages epistemic neural networks (Epinets) to estimate model uncertainty for every affinity inference, which is not available for existing models like Boltz-2 affinity.
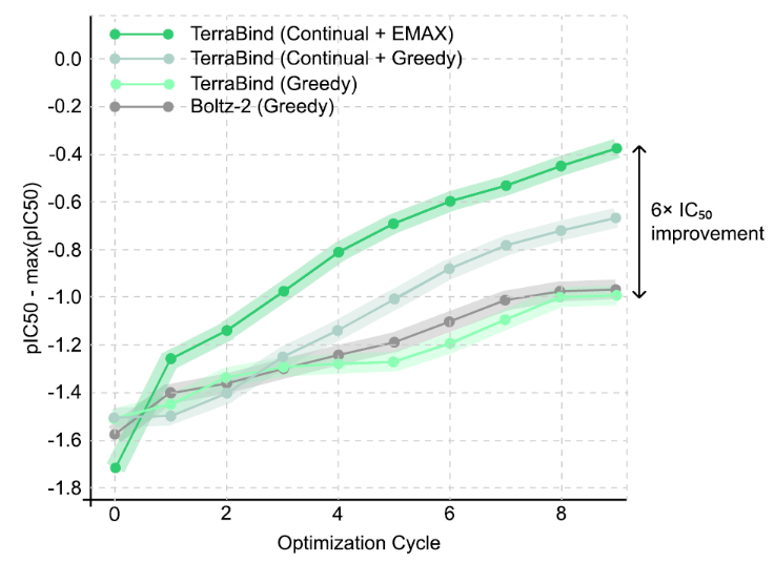
While these uncertainty estimates offer intrinsic value by adding perspective to individual predictions, the distinct advantage is that this allows TerraBind to predict joint affinity distributions across a batch of molecules. This enables the use of specialized acquisition functions like Expected Maximum (EMAX), which can better account for correlated uncertainties and diversity when making molecule selection decisions in practical Design-Make-Test-Analyze (DMTA) cycles. See our recent paper and associated blog for more info. Furthermore, TerraBind is amenable to continual learning, allowing rapid Bayesian conditioning on new experimental observations without the expense or complication of naive retraining or finetuning.
In our internal benchmark, this continual and EMAX-based approach yielded a 6x improvement in selection performance (maximizing IC50) compared to standard greedy methods.
What's Next?
At Terray, TerraBind is not an academic exercise. TerraBind-Seq and TerraBind-Struct are already central to our operations. We use these models continuously to drive decisions across our pipeline, allowing us to navigate massive chemical spaces with a level of speed and confidence that was previously unattainable. And we never rest, continually enhancing all of EMMI’s capabilities to more effectively deliver medicines to patients in need.